|
| | Nizatidine Chemical Properties |
| Melting point | 130-1320C | | Boiling point | 478.2±45.0 °C(Predicted) | | density | 1.249±0.06 g/cm3(Predicted) | | storage temp. | Keep in dark place,Inert atmosphere,Room temperature | | solubility | Sparingly soluble in water, soluble in methanol. | | form | neat | | pka | 2.1, 6.8(at 25℃) | | color | Off-White to Pale Yellow | | Water Solubility | 21.4g/L(temperature not stated) | | Merck | 14,6660 | | CAS DataBase Reference | 76963-41-2(CAS DataBase Reference) |
| Hazard Codes | Xn | | Risk Statements | 22 | | Safety Statements | 36 | | WGK Germany | 3 | | RTECS | KM6565000 | | HS Code | 29349990 | | Hazardous Substances Data | 76963-41-2(Hazardous Substances Data) | | Toxicity | LD50 in mice, rats (mg/kg): 265, >300 i.v.; 1685, 1680 orally (Pioch) |
| | Nizatidine Usage And Synthesis |
| Drugs inhibiting the secretion of gastric acid | Nizatidine is a drug successfully developed by Eli Lilly Company (United State) for inhibiting the gastric acid secretion. In 1987, it was first approved for entering into the market of the United States with the trade name “aixi”. Nizatidine, as a kind of potent H2 receptor antagonist, acts on the gastric acid secretion cells and blocks the formation of stomach acid, leading to the reduction of the gastric acid. It can also inhibit the gastric acid secretion resulting from food and chemical stimuli such as gastrin and acetylcholine. The potency is similar to that of ranitidine and superior to that of cimetidine. Clinical studies have shown that the drug can significantly inhibit gastric acid secretion at night for as long as 10 h. Under normal conditions, the drug does not affect the activity of pepsin in the gastric juice but may affect the secretion amount. It has no female antiandrogen effect. It causes no adverse reactions to the cardiovascular nerve and endocrine system, nor does affect the activity of hepatic cytochrome P450 oxidase. It has good oral absorption with the bioavailability being greater than 90% with PBP accounting for 30% and another 30% binding to the cells. t1/2 is about 1.3h with 90% to 100% excreted from the urine within 16h after the treatment, of which 65% to 75% is in the form of prototype, 6% is in the form N2-oxide, 8% is the N2-single demethylation compound. The saliva concentration is only about 1/3 of the plasma. The elimination half-life of the patients of renal failure patients is about 7h. Mild to moderate damage of the liver damage does not affect the metabolism of this product.
[Indications] it is suitable for the treatment of gastric and duodenal ulcers, gastroesophageal reflux disease and prevention of the recurrence of duodenal ulcer.
[Usage and dosage] Oral: stomach and duodenal ulcer; apply single administration of 300 mg before sleeping with the treatment course lasting for 4 to 8 weeks. You may also administer per time in both morning and evening with the dose of 150 mg each time; the maintenance dosage for the treatment of duodenal ulcer is 150 mg per night; for the treatment of gastroesophageal reflux disease, take 150 mg each time with 2 times per day and the treatment course should be more than 12 weeks; for prevention of ulcer, administer 150 mg once before sleeping.

Figure 1 The chemical formula of nizatidine.
| | The antagonist of the H2 receptor | H2 receptor antagonists are a class of effective gastric acid secretion inhibitors. Those drugs which currently have entered clinical application include cimetidine, ranitidine, famotidine, nizatidine and roxatidine. These drugs can competitively inhibit the binding of histamine with the H2 receptor of the parietal cells, thereby reducing the intracellular cAMP levels and gastric acid secretion. H2 receptor antagonists can also inhibit the fundamental gastric acid secretion as well as the gastric acid secretion occurring at night, but has no inhibitory effect on the gastric acid secretion stimulated by having meal. Administration of H2 receptor antagonists for 2 to 4 times daily can significantly increase the gastric pH of the nighttime but has only moderate effect on the daytime pH. For patients of peptic ulcer with no complication, the efficacy of taking twice daily is the same as that of taking once daily before sleeping. The recommended dose for the patients in the acute phase of peptic ulcer (once daily before sleeping) is: cimetidine: 800mg, ranitidine and nizatidine: 300 mg, famotidine: 40mg. The ulcer symptoms are usually completely alleviated within 2 weeks. The healing rate of duodenal ulcer in the 6 weeks to 8 weeks treatment course is about 85% to 90%. The healing of the stomach ulcer is two to four weeks later than that of the duodenal ulcer but when the treatment course is extended to 8 weeks, it can mostly get healed in most cases.
Patients have a well tolerance on the H2 receptor antagonists and rarely get serious side effects. The incidence of central nervous system symptoms such as headache, confusion and sleepiness is approximately 1%, particularly under intravenous infusion. Cimetidine can inhibit the metabolism of the P450 drug in the liver and can cause change on the serum concentration of drugs such as warfarin, theophylline, lidocaine and phenytoin. The ability of ranitidine in binding to P450 is one-tenth of that of cimetidine. Moreover, the effect of both famotidine and nizatidine effect is also very slight. Cimetidine can inhibit the metabolism of estradiol and testis acid dione which may lead to impotence and feminization of male breast. These cases are especially prevalent upon high-dose treatment.
The above information is edited by the chemicalbook of Dai Xiongfeng.
| | Uses | The value of nizatidine in the treatment of peptic ulcer disease is comparable with that of ranitidine and cimetidine. 1 or 2 times daily dosing can typically cure duodenal ulcer within 8 weeks; a single dose administration daily can prevent recurrence (Bianchi-Porro and Keohane, 1987; Stern, 1988). The efficacy of nizatidine in the treatment of stomach ulcers is similar as that of other H2-blockers but with little information available. For reflux esophagitis, Zollinger-Ellison syndrome and other kinds of peptic ulcer diseases, it is expected to generate a equal effect as ranitidine but this still needs clinical studies.
| | Side effects | This kind of drug can be well tolerated. It has been noted of some mild adverse reactions of the gastrointestinal tract. During the clinical trials, the elevated level of serum uric acid and transaminase observed in few patients are not clinically significant. Similar as other kinds of H2 receptor antagonists, the potential liver toxicity of applications of is low. Application of high dose to rats had ever produced anti-androgen effect, but this effect had not been observed in clinical trials of therapeutic doses (Stern, 1988). Nizatidine be classified as FDA pregnancy category C.
| | Drug Interactions | 1. This product can increase gastric pH, increase the midazolam absorption. Take caution when these two drugs are combined.
2. The product, when used in combination with ceftibuten, can increase the incidence of adverse reactions (such as nausea, diarrhea, headache) caused by the later one.
3. The product, if being used in combination with cefditoren, can reduce the plasma concentration of the latter one.
4. The product can increase gastric pH, resulting in decreased absorption of cefpodoxime and delavirdine.
5. This product can reduce the absorption of itraconazole, reducing its efficacy. When these two drugs are combined, the latter drug should be subject to oral administration together with cola drinks.
| | Precautions | 1. Patients allergic to this product, patients of liver and kidney dysfunction, pregnant women, lactating women and children should take with caution.
2. It can be used to cover the symptoms of stomach cancer, and can cause false positive to the urobilinogen determination.
| | Chemical Properties | Through crystallization from ethanol-ethyl acetate; it has a melting point of 130~132 ℃. Maximal UV absorption wavelength (methanol): 240,325nm (ε 8400, 19600); (water): 260,314nm (ε: 11820, 15790). pKal: 2.1: pKa2: 6.8. Partition coefficient (octanol/water): 0.3 (pH = 7.4). It is easily soluble in chloroform, soluble in methanol, slightly soluble in water or a buffer solution, slightly soluble in ethyl acetate or isopropyl alcohol and practically insoluble in benzene, diethyl ether, or octanol. Solubility (mg/ml): chloroform> 100.0, methanol: 50.0~100.0, water: 10.0~33.3 water, isopropanol: 3.33 to 5.0, ethyl acetate: 1.0 to 2.0, benzene, ether, and octanol: <0.5. Acute toxicity LD50: for mice, rats (mg/kg): 265, > 300 intravenously injection; oral 1685, 1680.
| | Uses | It is a kind of H2 receptor antagonists. It can be used for the prevention of active duodenal ulcer, benign gastric ulcer and prevention of recurrence after the healing of the active duodenal ulcer.
| | Production method | Nitromethane and carbon disulfide are put into absolute ethanol; add drop wise of the ethanol solution of potassium hydroxide at 33-35 ℃, filter after the completion of the reaction; the obtained red solid was suspended in methanol, add drop wise of iodomethane at 25 ℃ and continue for the reaction. The obtained yellow crude product was recrystallized with cyclohexanone to obtain golden yellow needles of 1, 1-dimethyl-2-nitroethylene with the yield being 68%. It is then react with cysteamine hydrochloride and potassium hydroxide in a mixture solution of ethanol and water for reflux to obtain the orange 2-nitromethylene-1, 3-thiazolidine with the yield being 65%. It is then further reacted with 33% methylamine ethanol solution at room temperature to give N-(2-mercaptoethyl)-N'-methyl-2-nitro-1, l-ethylene diamine with the yield being 85%.
33% aqueous solution of dimethylamine, sodium bisulfite and 36% formaldehyde are stirred together with water. Add potassium cyanide and continue the reaction to obtain dimethylamino acetonitrile with the boiling point being 34-38 ℃/2.7kPa and the yield being 83.6%. It is further mixed with anhydrous pyridine, dry triethylamine and anhydrous methanol, put through hydrogen sulfide for being heated at 80 ℃ and then stand at room temperature to obtain dimethylamino thioacetamide with the yield being 35%. Add it to the acetone solution of 1, 3-dichloroacetone at-5 °C, making 2-dimethylaminomethyl-4-chloro-4-hydroxy-methyl-thiazoline with the yield being 72%. Dissolve it in 1, 2-dichloroethane and add drop wise of the dichloroethane solution of thionyl chloride at room temperature. After the reaction at room temperature, further have reaction at 40 ℃ to give 2-dimethylaminomethyl-4-chloromethyl-thiazole hydrochloride with the yield being 62%. Finally, it is reacted with N-(2-mercaptoethyl)-N'-methyl-2-nitro-1,1-ethylene diamine reaction in aqueous solution of sodium hydroxide to produce nizatidine and recrystallized with methanol-ethyl acetate. The finished product has a melting point being 129-132 ℃ and the yield being 52%.
| | Description | Nizatidine is the fifth H2-antagonist introduced to the world market as an antiulcer agent.
It is reported to be effective in the treatment of both duodenal and gastric ulcers or the
prevention of their recurrences. Given once-daily, nizatidine’s bioavailability is not
diminished by the concurrent administration of an antacid. | | Chemical Properties | White Crystalline Powder | | Originator | Lilly (USA) | | Uses | Histamine H2-receptor antagonist related to Ranitidine (R120000). Antiulcerative. | | Uses | immune suppressant, antineoplastic, antiviral | | Uses | For the treatment of acid-reflux disorders (GERD), peptic ulcer disease, active benign gastric ulcer, and active duodenal ulcer. | | Uses | Nizatidine is used for treating stomach and duodenum
ulcers and other conditions accompanied by elevated acidity of the gastrointestinal tract. | | Definition | ChEBI: A member of the class of 1,3-thiazoles having a dimethylaminomethyl substituent at position 2 and an alkylthiomethyl moiety at position 4. | | Indications | Nizatidine is the newest H2-receptor antagonist.
Similar to ranitidine, it has a relative potency twice that
of cimetidine.About 90% of an oral dose is absorbed,
with a peak plasma concentration occurring after 0.5 to
3 hours; inhibition of gastric secretion is present for up
to 10 hours.The elimination half-life is 1 to 2 hours, and
more than 90% of an oral dose is excreted in the urine. | | Manufacturing Process | Nizatidine may be prepared by 2 ways.
1. A mixture of (25.7 g) 2-nitromethylenethiazolidine and acetonitrile (50 ml) was stirred and heated at 40°C under nitrogen. Methylamine gas (16.0 g) was passed into the stirred mixture over 45 minutes to give a solution. A slurry of 4-chloromethyl-2-dimethylaminomethylthiazole hydrochloride (40.0 g) (prepared as described in EP 49,618) in acetonitrile (50 ml) was added to the solution over a period of 4.5 hours whilst methylamine gas was bubbled through the reaction mixture such that methylamine (38.3 g) was added over the period (total methylamine added was 54.3 g). The temperature of the reaction mixture varied between 24° and 35°C during the addition. After the addition, the mixture was diluted with acetonitrile (50 ml) and stirred at ambient temperature for 17 hours. A solid was removed by filtration and the filtrate was split into 2 equal portions.
Portion 1: The solution was evaporated to give a black oil which was partitioned between water (200 ml) and chloroform (200 ml). The separated chloroform phase was washed with saturated brine, then dried over magnesium sulphate, filtered and evaporated to give a reddish oil which was dissolved in acetone (200 ml), boiled under reflux, cooled to 40°C and then seeded with nizatidine. The mixture was left to stand at 0°-5°C for 64 hours. The mixture was filtered to give nizatidine (10.4 g, 37%) m.p. 118-122°C. The structure was confirmed by1H NMR. The product was 95.4% pure by HPLC.
Portion 2: The mixture was evaporated to give an oil which was taken up in chloroform (200 ml) then washed with water (100 ml). The chloroform solution was washed with brine (100 ml), dried over magnesium sulphate, and then concentrated under reduced pressure at 45°C to give a brown oil. The oil was dissolved in acetone (200 ml) and activated charcoal (0.5 g) was added to the solution. The mixture was boiled under reflux for 10 minutes, then cooled to 45°C and filtered at this temperature to remove the charcoal. The filtrate was cooled to 20°C, seeded with nizatidine (0.05 g), then cooled 0°5°C for 45 minutes during which time crystallisation occurred. The mixture was filtered to give nizatidine (9.4 g, 32.2%).
2. A mixture of 2-nitromethylenethiazolidine (12.6 g) and water (30.0 ml) was stirred and heated at 40°C under argon. Methylamine (20.0 g of a 40% w/w aqueous solution) was added slowly over 30 minutes to the reaction mixture
at 40°C. The mixture was cooled at ambient temperature and further methylamine (23.6 g of 40% w/w aqueous solution) was added over 2.5 hours and a solution of 4-chloromethyl-2-dimethylaminomethylthiazole dihydrochloride (25.0 g) in water (30 ml) was added over 5.5 hours with the addition of the thiazole starting simultaneously with the addition of the methylamine. The reaction mixture was left to stir for a further 15 minutes and then was concentrated under reduced pressure. The solid obtained was dissolved in a mixture of methyl ethyl ketone (200 ml), aqueous potassium carbonate solution (43 ml, 10% w/w). The mixture was warmed slightly to obtain a solution. The mixture was separated and the aqueous layer was washed with methyl ethyl ketone (2 times 130 ml and then 1 times 100 ml). The combined organic layers were evaporated under reduced pressure to yield crude nizatidine (approximately 25.2 g), which was shown to be 89.4% pure by HPLC. The crude solid was dissolved in dichloromethane (300 ml). The solution was washed with water (3 times 75 ml). The combined aqueous layer and the washings were back extracted with dichloromethane and the combined organic layers were dried and concentrated under reduced pressure to give nizatidine (21.1 g, 74.3% yield). The solid was dissolved in ethanol (45 ml) by warming on a steam bath. The solution was removed from the steam bath treated with activated charcoal (2.3 g) and the mixture was boiled for a further 8 minutes. The mixture was hot filtered. The filtrate was cooled and filtered to give nizatidine (13.8 g, 48% yield) which was shown to be 99.8% pure by HPLC.
| | Brand name | Axid (Braintree); Axid (Reliant). | | Therapeutic Function | Antiulcer | | General Description | Nizatidine, N-[2-[[[2-(dimethylamino)methyl]-4-thiazolyl]methyl]thio]-ethyl]-N'-methyl-2-nitro-1,1-ethenediamine (Axid), is an off-white to buff crystallinesolid that is soluble in water, alcohol, and chloroform. It isa thaizole derivative of raniditine and has pKas of 2.1 (side chain) and 6.8 (dimethylamino). Nizatidine’s mechanism ofaction is similar to other H2-antagonists, as is its receptor selectivity.It is more potent than cimetidine.
Nizatidine has excellent oral bioavailability (>90%).The effects of antacids or food on its bioavailability arenot clinically significant. The elimination half-life is 1 to2 hours. It is excreted primarily in the urine (90%) andmostly as unchanged drug (60%). Metabolites include nizatidinesulfoxide (6%), N-desmethylnizatidine (7%), andnizatidine N-oxide (dimethylaminomethyl function).Nizatidine has no demonstrable antiandrogenic action, effectson other hormones, or inhibitory effects on cytochromeisozymes involved in the metabolism of other drugs. | | Clinical Use | H2
-receptor antagonist | | Synthesis | Nizatidine is N-[2-[[[2-[(dimethylamino)methyl]-4-thiazolyl]methyl] thio]
ethyl]-2-nitro-1,1-ethenediamine (16.2.15). According to its chemical structure, nizatidine
is somewhat of a hybrid structure of ranitidine and famotidine, in which a side chain of ranitidine
and carrying heterocycle, 2-aminothiazol, are used. Likewise, its synthesis also is a
specific combination of pathways used for making both prototype drugs. 2-(Dimethylaminomethyl)-
4-hydroxymethylthiazol serves as the initial compound, from which the
desired nizatidine (16.2.15) is synthesized by subsequent reaction with 2-mercaptoethylamine
hydrochloride and then with N-methyl-1-methythio-2-nitroethenamine. 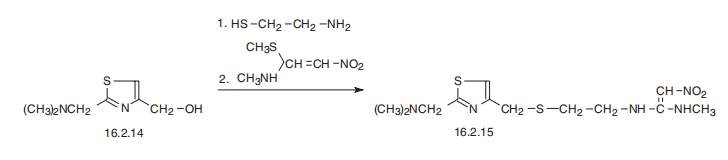
| | Veterinary Drugs and Treatments | While nizatidine acts similarly to cimetidine and ranitidine as an
H2 blocker to reduce gastric acid secretion in the stomach, in small
animal medicine its use has been primarily for its prokinetic effects.
It may be useful to treat delayed gastric emptying, pseudo-obstruction
of the intestine and constipation.
H2 blockers may be useful in preventing hemorrhagic necrosis
in feline pancreatitis. | | Drug interactions | Potentially hazardous interactions with other drugs
Antifungals: absorption of itraconazole, ketoconazole
and possibly posaconazole reduced, avoid with
posaconazole suspension.
Antivirals: concentration of atazanavir reduced;
concentration of raltegravir possibly increased -
avoid; avoid for 12 hours before and 4 hours after
rilpivirine.
Cytotoxics: avoid with dasatinib and erlotinib;
possibly reduced absorption of pazopanib - give at
least 2 hours before or 10 hours after nizatidine;
possibly reduced absorption of lapatinib.
Ulipristal: contraceptive effect possibly reduced -
avoid with high dose ulipristal. | | Metabolism | A small amount of nizatidine is metabolised in the liver:
nizatidine N-2-oxide, nizatidine S-oxide, and N-2-
monodesmethylnizatidine have been identified, the latter
having about 60% of the activity of nizatidine.
More than 90% of a dose of nizatidine is excreted in the
urine, in part by active tubular secretion, within 12 hours,
about 60% as unchanged drug. Less than 6% is excreted in
the faeces |
| | Nizatidine Preparation Products And Raw materials |
|


