|
| | Fondaparinux sodium Basic information |
| Product Name: | Fondaparinux sodium | | Synonyms: | Fondaparin SodiuM;Methyl O-2-Deoxy-6-O-sulfo-2-(sulfoaMino)-α-D-glucopyranosyl-(14)-O-β-
D-glucopyranuronosyl-(14)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoaMino)-α-D-glucopyranosyl-(14)-O-2-O-sulfo-α-L-idopyranuronosyl-(14)-2-deoxy-2-(sulfoaMino)-α-D-glucopyranoside 6-(Hydrogen Sulfate) DecasodiuM Salt;Fondaparinux SodiuM Identification;Methyl O-2-Deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(14)-O-β- D-glucopyranuronosyl-(14)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(14)-O-2-O-sulfo-α-L-idopyranuronosyl-(14)-2-deoxy-2-(sulfoamino)-α-D-glucopyra;Fondaparinux sodium N-4;Fondaparinux sodium, >=98%;decasodium 6-[[6-[[2-carboxylato-4-hydroxy-6-[[4-hydroxy-6-methoxy-5-(sulfonatoamino)-2-(sulfonatooxymethyl)-3-oxanyl]oxy]-5-sulfonatooxy-3-oxanyl]oxy]-5-(sulfonatoamino)-4-sulfonatooxy-2-(sulfonatoox;decasodium (2R,3S,4S,5R,6R)-3-[(2R,3R,4R,6R)-5-[(2R,3R,4S,5S,6S)-6-carboxylato-5-[(2R,3R,4R,5S,6R)-4,5-dihydroxy-3-(sulfonatoamino)-6-(sulfonatooxymethyl)oxan-2-yl]oxy-3,4-dihydroxy-oxan-2-yl]oxy-3-(sulfonatoamino)-4-sulfonatooxy-6-(sulfonatooxymethyl)oxan-2-yl]oxy-4-hydroxy-6-[(2R,3S,4R,5R,6S)-4-hydroxy-6-methoxy-5-(sulfonatoamino)-2-(sulfonatooxymethyl)oxan-3-yl]oxy-5-sulfonatooxy-oxane-2-carboxylate | | CAS: | 114870-03-0 | | MF: | C31H43N3O49S8.10Na | | MW: | 1728.08 | | EINECS: | 686-283-7 | | Product Categories: | Intermediates & Fine Chemicals;Oligosaccharides;Pharmaceuticals;Sulfur & Selenium Compounds;114870-03-0 | | Mol File: | 114870-03-0.mol |  |
| | Fondaparinux sodium Chemical Properties |
| Melting point | >209°C (dec.) | | alpha | D23 +48° (c = 0.61 in water) | | storage temp. | 2-8°C | | solubility | Water | | form | Solid | | color | White to Off-White | | InChIKey | XEKSTYNIJLDDAZ-UWCYYFDCNA-D |
| | Fondaparinux sodium Usage And Synthesis |
| Description | Fondaparinux sodium was first introduced in the US for prophylaxis of deep vein
thrombosis which may lead to pulmonary embolism following major orthopaedic surgery.
Fondaparinux is the first of a new class of antithrombic agents distinct from low molecular
weight heparin (LMWH) and heparin. This entirely synthetic molecule is a copy of the
heparin pentasaccharide sequence, the shortest fragment able to catalyze antithrombin lllmediated
inhibition of factor Xa thereby inhibiting thrombin generation without antithrombin
action. Fondaparinux does not display significant effects on coagulation tests (such as activated partial thromboplastin time and prothrombin time), does not bind to platelet factor
4 or promote heparin-induced thrombocytopenia. In phase III studies, fondaparinux
significantly reduced the incidence of thromboembolism following orthopedic surgery, with
an overall risk reduction of 50% in comparison to the LMWH, enoxaparin. Following
subcutaneous administration, fondaparinux has a nearly complete bioavailability, a rapid
onset of action, a prolonged half-life (17.2 h) enabling once daily dosing and is not
metabolized preceeding renal excretion. The drug appears to be generally safe, with
haemoragic complications either comparable to or higher than those for LMWH. | | Chemical Properties | White Powder (after lyophilisation) | | Originator | Sanofi-Synthelabo (France) | | Uses | Fondaparinux sodium has been used to test its neutralizing effect towards enterovirus D68-947 infection. It may be used in ultraviolet photodissociation (UVPD) measurements. | | Uses | Synthetic pentasaccharide corresponding to the anti-thrombin binding site of heparin. Anti-thrombotic. | | Definition | ChEBI: An organic sodium salt, being the decasodium salt of fondaparinux. | | Brand name | Arixtra (GlaxoSmithKline). | | Biochem/physiol Actions | Fondaparinux sodium is an antithrombotic anticoagulant, a Factor Xa inhibitor. Fondaparinux sodium is chemically related to low molecular weight heparins. Its pentasaccharide structure corresponds to the antithrombin III (ATIII) binding site of heparin. Fondaparinux sodium binding at this site potentiates the natural inhibitory effect of ATIII against factor Xa by a factor of approximately 300, which results in inhibition of thrombin generation. | | Clinical Use | Prophylaxis of deep vein thrombosis
Treatment of deep vein thrombosis, pulmonary
embolism, unstable angina and after a myocardial
infarction | | Synthesis | Starting from Dglucose,
D-cellobiose, and D-glucosamine, the production
process for the synthesis of the pentasaccharide involves
about 55 steps. The synthesis was accomplished by
preparing a fully-protected pentasaccharide, and then
converting it into the final product. The choice of protecting
groups was dictated by two factors: the need to introduce
sulfate substituents (O- as well as N-linked), carboxylate
groups and hydroxyl groups, in the proper positions on the
target molecule, and the constraints of current methods for
oligosaccharide synthesis, particularly the use of 2-azido
glucose derivatives to achieve stereoselective introduction of
|á-D-linked glucosamine units. All the monosaccharide
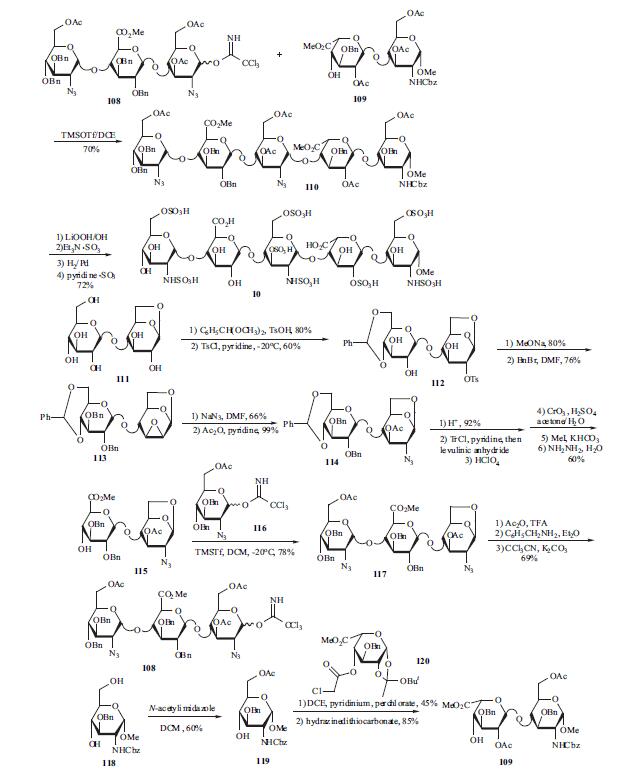
synthons were obtained from glucose or from glucosamine, and the synthesis is outlined in the scheme.
Trisaccharide 108 and disaccharide 109 are the two key
building blocks in the synthesis. Coupling 108 and 109 was
carried out at -20??C in DCE. Fully protected pentasaccharide
110 was then converted into the target compound 10 using
traditional methods: saponification, O-sulfation, cleavage of
benzyl ethers with simultaneous reduction of azido into amino functions and finally N-sulfation. Preparation of
trisaccharide building block 108 started from 1,6-anhydrocellobiose
(111). Selective protection at 4?ˉ,6?ˉ position was
achieved through benzylidenation to provide crude 112
which was converted into epoxide 113 by treatment with
sodium methoxide and benzylation. Compound 113 was isolated after filtration on silica gel and crystallization (m.p.
184-5??C). Trans-diaxial opening of the epoxide yielded the
2-azido derivative (66%) which was acetylated to give 114
(99%). The benzylidene was cleaved (92%) and the diol was
then converted into 115 by successive tritylation,
levulinoylation, detritylation, oxidation, methylation and hydrazinolysis (60% over the 6 steps). Imidate 116 was
prepared in the usual way from its hydroxyl precursor and
coupled with 115 to give O-linked trisaccharide 117 in 78%
yield. Compound 117 was acetolysed (91%), the anomeric
acetate was cleaved by benzylamine in ether (100%) and
imidate 108 was obtained by reaction with potassium
carbonate and trichloroacetonitrile at room temperature (|á,
|?- mixture with |á as the predominant isomer, 76%). The
preparation of the other building block 109 is described as
following. Selective 6-acetylation of 118 by N -
acetylimidazole in DCE gave 119 in 60% yield. Treatment
of 119 with 120 using DCE/pyridinium perchlorate and
followed dechloroacetylation using hydrazinedithiocarbonate
afforded the crystalline disaccharide 109.
| | Drug interactions | Potentially hazardous interactions with other drugs
Increased risk of bleeding in combination with any
other drugs that affect coagulation. | | Metabolism | Although not fully evaluated, there is no evidence of
fondaparinux metabolism and in particular no evidence
for the formation of active metabolites.
Fondaparinux is excreted to 64-77% by the kidney as
unchanged compound. |
| | Fondaparinux sodium Preparation Products And Raw materials |
|

