|
| | Doripenem Basic information |
| Product Name: | Doripenem | | Synonyms: | (+)-(4r,5s,6s)-6-[(1r)-1-hydro-xyethyl]-4-methyl-7-oxo-3-[[(3s,5s)-5-[(sulfamoylamino)-methyl]-3-pyrrolidinyl]thio]-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid;DORIPENEM;Doripenem/(+)-(4R,5S,6S)-6-[(1R)-1-Hydro-xyethyl]-4-methyl-7-oxo-3-[[(3S,5S)-5-[(sulfamoylamino)-methyl]-3-pyrrolidinyl]thio]-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid;(4R,5S,6S)-3-[[(3S,5S)-5-[[(Aminosulfonyl)amino]methyl]-3-pyrrolidinyl]thio]-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic Acid;S 4661;(4R,5S,6S)-6-((R)-1-hydroxyethyl)-4-Methyl-7-oxo-3-((3S,5S)-5-((sulfaMoylaMino)Methyl)pyrrolidin-3-ylthio)-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid;InterMediates of DoripeneM;DoripeneM Monohydrate | | CAS: | 148016-81-3 | | MF: | C15H24N4O6S2 | | MW: | 420.5 | | EINECS: | 1308068-626-2 | | Product Categories: | Doribax, Doripenem monohydrate, S-4661, Finibax;Chiral Reagents;Intermediates & Fine Chemicals;Pharmaceuticals;Sulfur & Selenium Compounds;intermediates | | Mol File: | 148016-81-3.mol |  |
| | Doripenem Chemical Properties |
| Melting point | >186°C dec. | | Boiling point | 694.8±65.0 °C(Predicted) | | density | 1.59±0.1 g/cm3(Predicted) | | storage temp. | Sealed in dry,Store in freezer, under -20°C | | solubility | In water, not known, in DMSO 20 mg/ml, in pBS 3 mg/ml | | pka | 4.27±0.60(Predicted) | | form | Solid | | color | White to Light Beige | | Stability: | Hygroscopic | | CAS DataBase Reference | 148016-81-3 |
| | Doripenem Usage And Synthesis |
| Description | Doripenem monohydrate is an ultra-broad-spectrum injectable β-lactam antibiotic and belongs to the subgroup of carbapenems. It was introduced by Shionogi Co. of Japan under the brand name Finibax in 2005 and is being marketed outside Japan by Johnson & Johnson. Doripenem act by decreases the process of cell wall growth, which eventually leads to elimination of the infectious cell bacteria together. It is used for treatment of bacterial respiratory and urinary tract infections. Doripenem is a 1β-methyl carbapenem derivative, and it is the fourth analog to be marketed in this series following the launch of meropenem, biapenem, and ertapenem in previous years. The introduction of a 1β-methyl group to the carbapenem skeleton enhances metabolic stability to renal dehydropeptidase-1 (DHP-1) and leads to improved antibacterial potency. | | Chemical Properties | Doripenem white to somewhat yellowish crystalline powder which is moderately soluble in water, slightly soluble in methanol, and virtually insoluble in ethanol. Doripenem is also solution in N,N-dimethylformamide. The chemical configuration of doripenem’s has 6 asymmetrical carbon atoms (6 stereocenters) and is most commonly supplied as one pure isomer. In terms of doripenem for injection, the crystallized powered drug can form a monohydrate when mixed with water. | | Originator | Shinogi (Japan) | | Uses | Doripenem hydrate is used to treat complicated urinary infection including Pyelonephritis caused by E.coli. Doripenem hydrate is promoted in the United States as DORIBAX(R). This drug is synthesized from p-nitrobenzyl-protected enolphosphate 2b and N-(p-nitrobenzyloxycarbonyl)-protected aminomethylpyrrolidine. | | Definition | ChEBI: Doripenem is a member of carbapenems. | | Brand name | Doribax,Finibax | | Antimicrobial activity | Doripenem have a broad spectrum of bacterial activity including both gram-positive and gram-negative bacteria but it is not active against MRSA. It is stable against β-lactamases including those with extended spectrum, but it is susceptible to the action of carbapenemases (Mandell, 2009). Thus it can be used in the treatment of infections such as: complex abdominal infections, pneumonia within the setting of a hospital, and complicated infections of the urinary tract including kidney infections with septicemia. Doripenem is also more active against Pseudomonas aeruginosa then other carbapenems. | | Pharmacokinetics | Cmax 500 mg intravenous infusion (1 h): c. 23 mg/L after 1 h
500 mg intravenous infusion (4 h): c. 8 mg/L
Plasma half-life: 1 h
Volume of distribution: 16.8 L (steady state)
Plasma protein binding: 8.1%
Absorption and distribution
Doripenem is not absorbed after oral administration. It penetrates well into most tissues and fluid, achieving concentrations matching or exceeding those required to inhibit most susceptible bacteria at the site of infection for the approved indications.
Metabolism and excretion
Metabolism of doripenem to the microbiologically inactive ring-opened metabolite occurs primarily by renal dehydropeptidase. Based on area under the concentration–time curve (AUC) values in plasma following a single 500 mg dose in healthy volunteers, 18% appears as metabolite and the rest as unchanged drug.
Excretion is primarily by the renal route. Within 24 h after dosing, 78.7% and 18.5% of the dose was recovered in urine as unchanged drug and the ring-opened metabolite, respectively. After administration of radiolabeled doripenem, 0.7% of the total radioactivity was recovered in feces after 1 week. | | Clinical Use | Doripenem is indicated for use for the treatment of intra-abdominal infections, and complicated urinary tract infections.
Complicated urinary tract infections, including pyelonephritis
Nosocomial pneumonia, including ventilator-associated pneumonia (Europe) | | Side effects | The most commonly reported adverse effects of doripenem include pain or swelling at the injection site, nausea, headache, and diarrhea.
Seizure and central nervous system (CNS) side effects are observed rarely (<1%), though headache is reported by 2.3% of patients. Other common drug-related adverse reactions are diarrhea (2.0%), nausea (1.9%), anemia (1.4%) and phlebitis (1.4%). Hypersensitivity reactions related to intravenous administration of the study drug and Clostridium difficile colitis occurred at a rate of less than 1%. However, patients with a history of hypersensitivity reactions to other β-lactam agents should be treated cautiously. | | Synthesis | the hydroxyl proline is protected as the PNZ ester 32 first in
95% yield. The protected proline acid 32 was converted to
the methyl ester with refluxing sulfuric acid in methanol
followed by conversion of the alcohol to the mesylate 34 in
91% overall yield from 30. The mesylate ester was reduced
with sodium borohydride to provide alcohol 35, which was
converted without purification to thiol ester 36 by reacting
with potassium thioacetate. Mitsunobu reaction
of alcohol 36 with BOC-sulfonyl urea 38, which was prepared
from chlorosulfonyl isocyanate with ammonia in tbutanol
in 90% yield, provided the key thioacetate intermediate
39. Finally, protected doripenem 42 was prepared by
coupling thiol 40, obtained by hyrolysis of thioacetate 39,
with enolphosphate 41 in 88% yield. Deprotection
of intermediate ester and carbamate protecting groups
via hydrogenation gave the desired carbapenem VI, which
was isolated after crystallization. Final form of the drug
doripenem was prepared by sterilization, crystallization and
granulation. 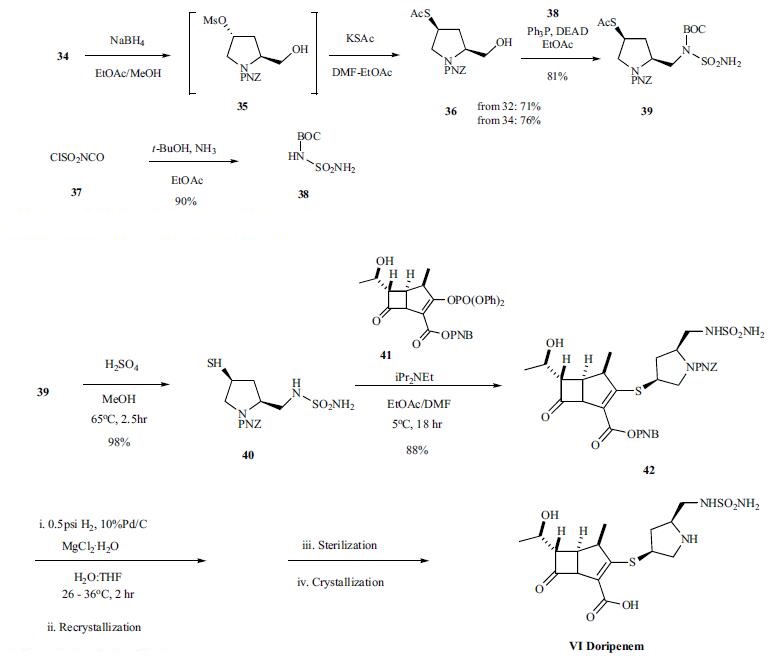
| | Dosage | Doripenem was initially developed in Japan and has received approval there in 2005 as Finibax. Finibax is indicated for multiple bacterial infections, the majority of which are respiratory indications.
The usual dose, according to the Japanese label, is 250 mg intravenously (i.v.) infused over 30 to 60 minutes 2 or 3 times a day; the maximum dose is 500 mg per administration up to a total dose of 1,500 mg/day. | | Mode of action | Doripenem's bactericidal function is due to its inhibition of the third stage of bacterial cell wall synthesis. Binding to penicillin-binding proteins weakens the cell wall and leads to cell death due to lysis of the cell wall.
Doripenem is effective against both grampositive and gramnegative aerobic bacteria.It may be more potent in vitro against Pseudomonas aeruginosa than meropenem. |
| | Doripenem Preparation Products And Raw materials |
|

