|
| | Clevudine Basic information |
| Product Name: | Clevudine | | Synonyms: | 1-[(2S,3R,4S,5S)-3-fluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl]-5-methyl-pyrimidine-2,4-dione;CLEVUDINE;1-(2'-DEOXY-2'-FLUORO-BETA-L-ARABINOFURANOSYL)-5-METHYLURACIL;1-[(2S,3R,4S,5S)-3-Fluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-methylpyrimidine-2,4-dione;2'-Fluoro-5-methylarabinosyluracil;Levovir;1-(2''-DEOXY-2''-FLUORO--L-ARABINOFURANOSYL)-5-METHYLURACIL;CLEVUDINE: 1-(2''-DEOXY-2''-FLUORO-BETA-L-ARABINOFURANOSYL)-5-METHYLURACIL | | CAS: | 163252-36-6 | | MF: | C10H13FN2O5 | | MW: | 260.22 | | EINECS: | 200-001-2 | | Product Categories: | Inhibitors;API;Bases & Related Reagents;Intermediates & Fine Chemicals;Nucleotides;Pharmaceuticals | | Mol File: | 163252-36-6.mol |  |
| | Clevudine Chemical Properties |
| Melting point | 184-185° | | alpha | D25 -111.77° (c = 0.23 in methanol) | | density | 1.55±0.1 g/cm3(Predicted) | | storage temp. | under inert gas (nitrogen or Argon) at 2-8°C | | solubility | DMSO (Slightly), Methanol (Slightly) | | pka | 9.55±0.10(Predicted) | | form | Solid | | color | White | | CAS DataBase Reference | 163252-36-6 |
| Toxicity | LD50 in mice, rats (mg/kg): >5000, >3000 orally (Painter) |
| | Clevudine Usage And Synthesis |
| Description | Clevudine is a fluorinated b-L-nucleoside analog launched for the
oral treatment of chronic HBV infection. It is the fifth nucleoside or nucleotide
analog to be marketed for this indication. The previous drugs from this class
include lamivudine, adefovir, entecavir, and telbivudine. In HBV-expressing
human hepatoma cell line 2.2.15, clevudine inhibits HBV DNA synthesis with
an EC50 of 0.1 μM, and does not show cytotoxicity up to 200 μM. It is
phosphorylated by cellular kinases to the active triphosphate derivative,
which subsequently inhibits HBV DNA polymerase and HBV replication.
466 Shridhar Hegde and Michelle Schmidt
Clevudine-5′-triphosphate has an intracellular half-life of 16.5 h. Interestingly, it
is a non-competitive inhibitor of viral polymerase, and inhibits HBV replication
without being incorporated into the DNA. The pharmacokinetic profile of clevudine was linear with a plasma half-life of approximately 60 h. Clevudine was undetectable in plasma after 4 weeks following the cessation of dosing.The most common adverse events reported with clevudine treatment include infection, asthenia, dyspepsia, abdominal pain, headache, and diarrhea.
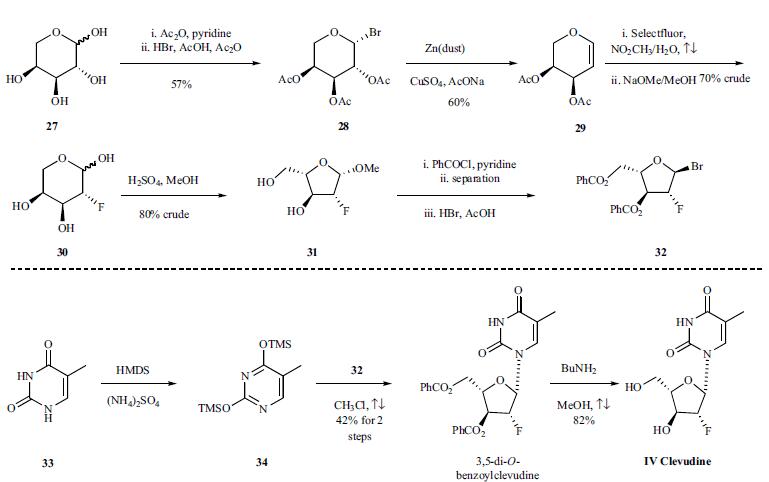
Clevudine is chemically derived from L-ribose by first incorporating acyl protective groups to produce 1-O-acetyl-2,3,5-tri-O-benzoyl-b-L-ribofuranose intermediate, which is then converted to 1,3,5-tri-O-benzoyl-a-L-ribofuranose in two steps by treating To Market, To Market 2007 467 with hydrogen chloride and subsequent hydrolysis and acyl migration. The remaining steps leading to clevudine include conversion of the C2-hydroxy group to C2-fluoro group with triethylamine trihydrofluoride, formation of the corresponding ribofuranosyl bromide intermediate with hydrogen bromide and acetic acid, condensation with silylated thymine, and removal of benzoyl protective groups with methanolic ammonia. | | Chemical Properties | White Solid | | Originator | University of Georgia/
Yale University (US) | | Uses | Antiviral drug, used for the treatment of Hepatitis B. | | Definition | ChEBI: Clevudine is a pyrimidine 2'-deoxyribonucleoside. | | Brand name | Levovir | | Synthesis | The synthesis is depicted in
the scheme. L-Arabinose (27) was treated with acetic
anhydride and pyridine at room temperature for four hours to give acetylated arabinose, which was then brominated using
30% HBr in AcOH/Ac2O at room temperature for 36 hours
to afford bromo-sugar 28 as a white solid in 57% yield after
crystallization in ethyl ether. Bromo-sugar 28 was then
treated with Zn dust, CuSO4 and NaOAc in AcOH/H2O, followed
by chromatographic separation to give L-arabinal 29
in 60% yield. L-arabinal 29 was converted to the fluoro derivative
in 70% crude yield by reaction with Selectfluor® (FTEDA-
BF4) in refluxing nitromethane/H2O, and the resulting
fluoroalcohol was deacetylated with NaOMe in MeOH to
give compound 30 in 100% crude yield. Compound 30 was
then treated with H2SO4 in refluxing MeOH to afford methyl
furanoside 31 in 80% crude yield. Furanoside 31 was benzoylated
with benzoyl chloride in pyridine to give a mixture
of isomers, from which the |á-anomer was isolated by chromatography
and then brominated with 30% HBr/AcOH in
CH2Cl2 to provide the crude bromo-sugar 32 which was dissolved
in chloroform and used without further purification in
the next step. Compound 34 was obtained by treatment of
thymine (33) with HMDS and ammonium sulfate in refluxing
chloroform for 16 hours. The sugar 32 was condensed with
silylated pyrimidine derivative 34 in refluxing chloroform to
afford 3,5-di-O-benzoylclevudine in 42% yield after recrystallization
from ethanol. The benzoyl groups were removed
upon treatment with n-butylamine in refluxing
methanol to give clevudine (IV) in 82% yield.
|
| | Clevudine Preparation Products And Raw materials |
|

